








































高斯计算(Gaussian)
计算样例
1) 氢化物中的键焓和静电势
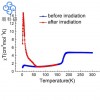
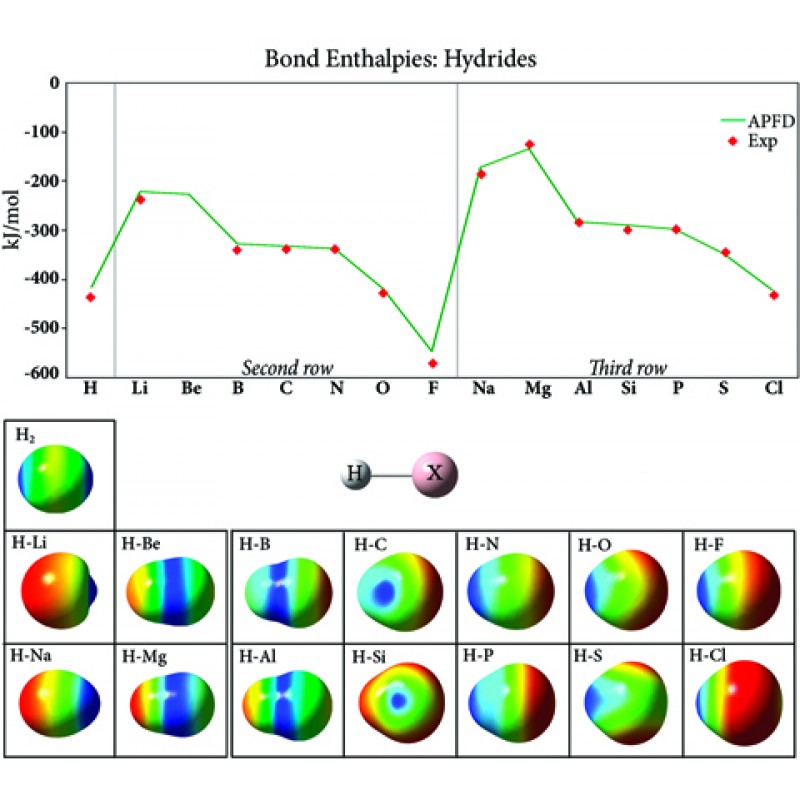
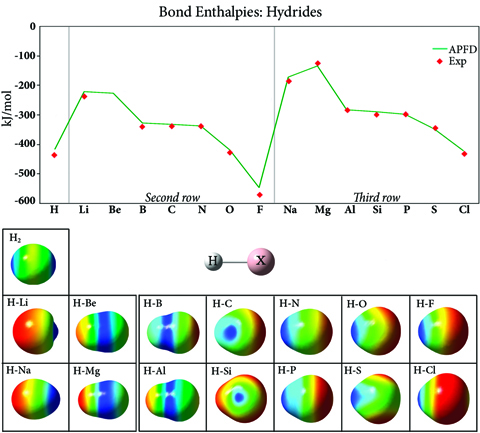 该图绘制了第二和第三行氢化物化合物中的键强度,通常在整个周期表中增加,最强键出现在惰性气体之前的元素中。该图的两行整体形状相似,但第三行的值更高,这是由于填充的第二个壳对细胞核提供了额外的屏蔽。图像显示了映射到等密度表面上的每种化合物的静电势。H2表面说明了这种键的共价性质;其他氢化物化合物中的键是离子键。负静电势(红色)位于每行开头的氢原子上,随着行内原子序数的增加,它移动到取代基上。因此,由于电负性的变化,氢化物键强度在一个周期(行)中增加,并随着您向下一个组(列)而减少。
2) 星际空间中的C60
该图绘制了第二和第三行氢化物化合物中的键强度,通常在整个周期表中增加,最强键出现在惰性气体之前的元素中。该图的两行整体形状相似,但第三行的值更高,这是由于填充的第二个壳对细胞核提供了额外的屏蔽。图像显示了映射到等密度表面上的每种化合物的静电势。H2表面说明了这种键的共价性质;其他氢化物化合物中的键是离子键。负静电势(红色)位于每行开头的氢原子上,随着行内原子序数的增加,它移动到取代基上。因此,由于电负性的变化,氢化物键强度在一个周期(行)中增加,并随着您向下一个组(列)而减少。
2) 星际空间中的C60
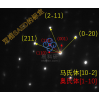
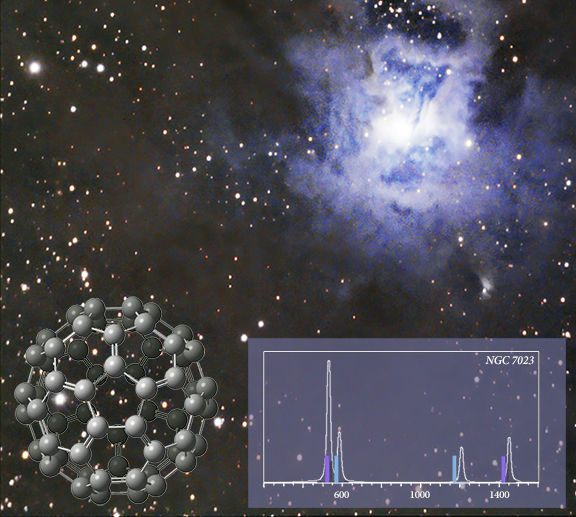 2004 年,在虹膜星云(NGC 7023) 的红外观测中发现了C60 [ Werner04,Sellgren10 ]。插图显示了从叠加在 APFD/6-311+G(2d,p) 模型化学预测的光谱上的数据(实心条)确定的峰位置。最强的峰(紫色)与实验室红外光谱相差 0.03-0.06 μm。
3) 有机磷农药的热化学
2004 年,在虹膜星云(NGC 7023) 的红外观测中发现了C60 [ Werner04,Sellgren10 ]。插图显示了从叠加在 APFD/6-311+G(2d,p) 模型化学预测的光谱上的数据(实心条)确定的峰位置。最强的峰(紫色)与实验室红外光谱相差 0.03-0.06 μm。
3) 有机磷农药的热化学
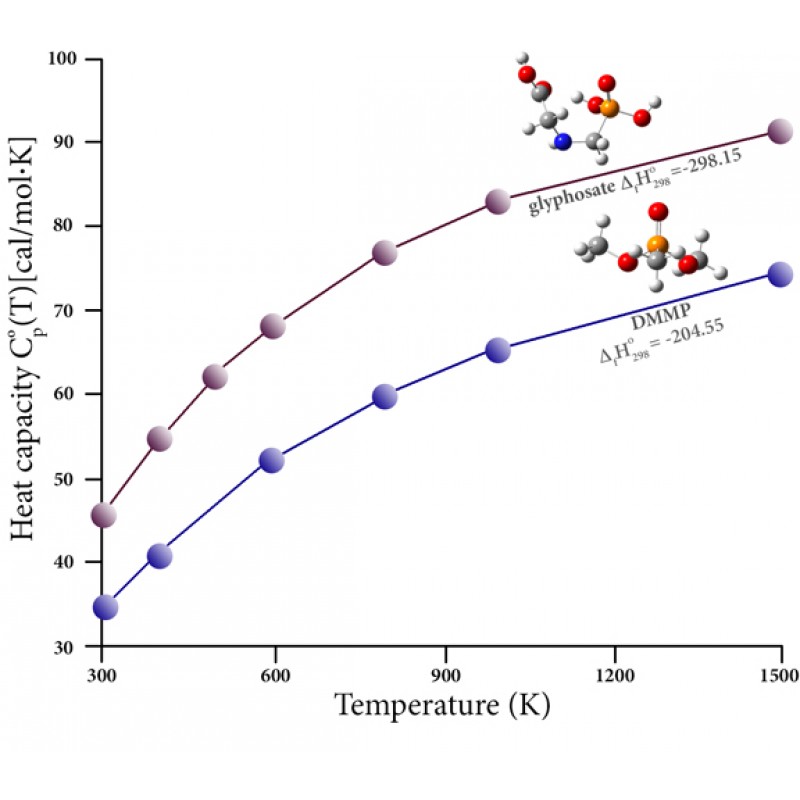
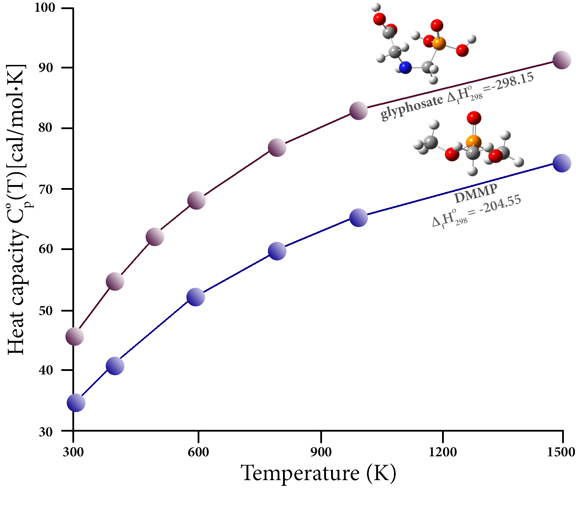 有机磷化合物通常用作杀虫剂(在许多其他应用中)。这些化合物由于其固有的毒性和燃烧过程中产生的有害产物(例如,由于燃烧先前处理过的植物材料)而对人类健康产生不利影响。这些化合物的分解很难通过实验研究;它们的热化学数据很少。然而,高精度的热化学预测可以弥补这一差距,并允许研究相关化合物和燃烧产物的热稳定性。例如,该图绘制了两种此类化合物的热容与温度的函数关系:农药草甘膦和更温和的阻燃化合物甲基膦酸二甲酯 (DMMP)。它还报告了它们的形成热(千卡/摩尔),哈尔法[ 15 ]。他们的论文提供了大量三价和五价磷化合物的计算结果,这些数据使他们能够提出 83 个原始组用于 Benson 的半经验群贡献方法,从而使他们能够评估一些常见的热化学性质。杀虫剂、除草剂和相关化合物。
4) [UO2(NC)4]2–和[UO2(CN)4]2-的最高占用 MO
有机磷化合物通常用作杀虫剂(在许多其他应用中)。这些化合物由于其固有的毒性和燃烧过程中产生的有害产物(例如,由于燃烧先前处理过的植物材料)而对人类健康产生不利影响。这些化合物的分解很难通过实验研究;它们的热化学数据很少。然而,高精度的热化学预测可以弥补这一差距,并允许研究相关化合物和燃烧产物的热稳定性。例如,该图绘制了两种此类化合物的热容与温度的函数关系:农药草甘膦和更温和的阻燃化合物甲基膦酸二甲酯 (DMMP)。它还报告了它们的形成热(千卡/摩尔),哈尔法[ 15 ]。他们的论文提供了大量三价和五价磷化合物的计算结果,这些数据使他们能够提出 83 个原始组用于 Benson 的半经验群贡献方法,从而使他们能够评估一些常见的热化学性质。杀虫剂、除草剂和相关化合物。
4) [UO2(NC)4]2–和[UO2(CN)4]2-的最高占用 MO
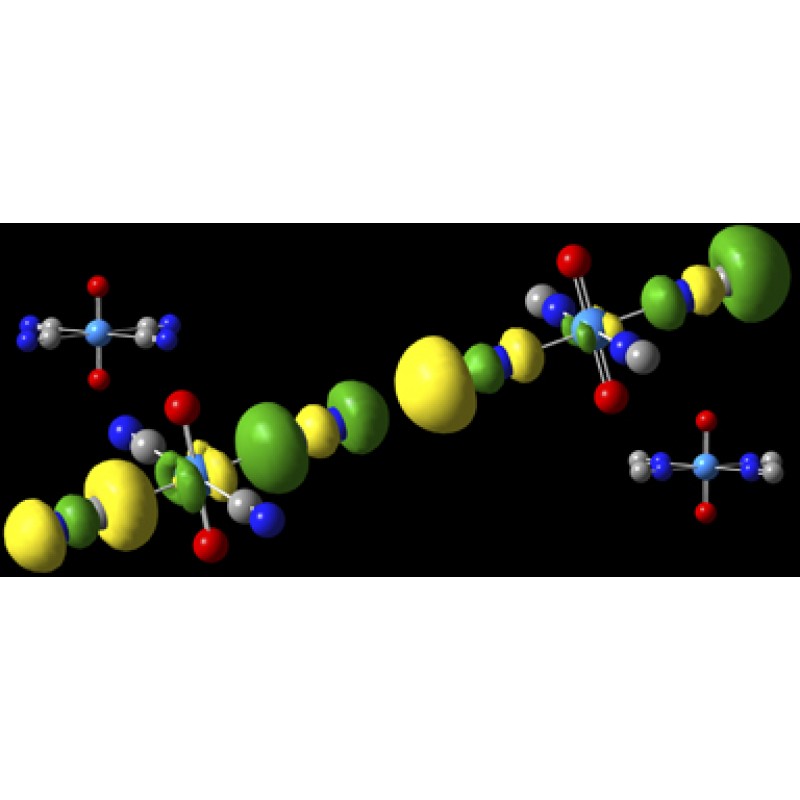
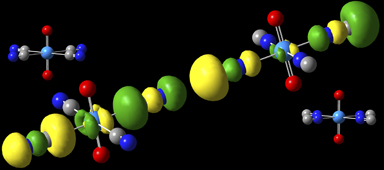 这些化合物使用B3LYP/aug-cc-pVDZ模型化学建模,使用Stuttgart/Dresden ECP和基于铀的[7s6p5d3f]基础设置[Sonnenberg05]。通常,与NC-复合物(右)相比,MO在CN-复合物(左)中更加稳定。例如,在HOMO中,位于碳原子上的5σ轨道在前者中指向铀原子,而在后者中指向铀原子,并且该基团与U的键长在CN中长0.13Å-复杂的。
5) 17O过渡金属配合物中的化学位移
这些化合物使用B3LYP/aug-cc-pVDZ模型化学建模,使用Stuttgart/Dresden ECP和基于铀的[7s6p5d3f]基础设置[Sonnenberg05]。通常,与NC-复合物(右)相比,MO在CN-复合物(左)中更加稳定。例如,在HOMO中,位于碳原子上的5σ轨道在前者中指向铀原子,而在后者中指向铀原子,并且该基团与U的键长在CN中长0.13Å-复杂的。
5) 17O过渡金属配合物中的化学位移
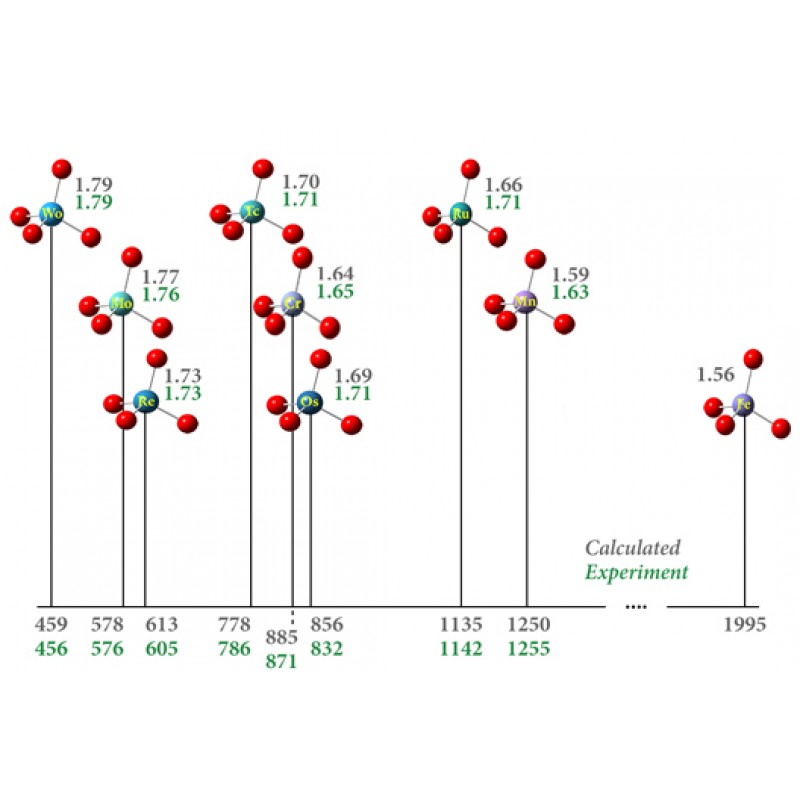
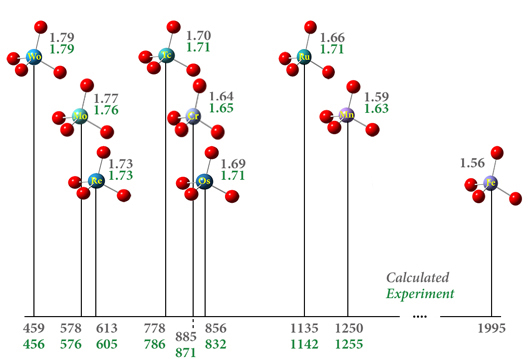 该图报告了一系列过渡金属配合物(灰色值)相对于水蒸气 (ppm) 和氧取代基键长 (Å) 的预测化学位移。这些化合物使用 APFD 泛函与 def2-TZVPP 基组和金属原子上的 ECP 建模。结果与实验非常吻合
该图报告了一系列过渡金属配合物(灰色值)相对于水蒸气 (ppm) 和氧取代基键长 (Å) 的预测化学位移。这些化合物使用 APFD 泛函与 def2-TZVPP 基组和金属原子上的 ECP 建模。结果与实验非常吻合
主要功能
Gaussian软件能够研究诸多的科学问题: (1)化学反应过程,如稳态及过渡态结构确定、反应热、反应能垒、反应机理及反应动力学等; (2)各类型化合物稳态结构的确定,如中性分子、自由基、阴、阳离子等 (3)各种谱图的验证及预测,如IR, Raman, NMR, UV/Vis, VCD, ROA, ECD, ORD, XPS, EPR, Franck-Condon及超精细光谱等; (4)分子各种性质,如静电势、偶极矩、布居数、轨道特性、键级、电荷、极化率、电子亲和能、电离势、自旋密度、电子转移、手性等; (5)热力学分析,如熵变、焓变、吉布斯自由能变、键能分析及原子化能等; (6)分子间相互作用,如氢键及范德华作用; (7)激发态,如激发态结构确定、激发能、跃迁偶极矩、荧光光谱、磷光光谱、势能面交叉研究等
收费标准
根据计算具体需求评估计算工期和费用,详情请联系在线客服
高斯计算(Gaussian)
Gaussian软件是目前计算化学领域内最流行、应用范围最广的综合性量子化学计算程序包。Gaussian软件基于量子力学而开发,它致力于把量子力学理论应用于实际问题,它可以通过一些基本命令验证和预测目标体系几乎所有的性质。此外,可视化软件GaussView的发布及计算机的快速发展更是大大降低了理论计算的门槛,使得各领域研究者能够轻松使用Gaussian研究和分析各种科学问题。
![]()