







































第一性原理DFT计算
计算样例
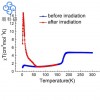
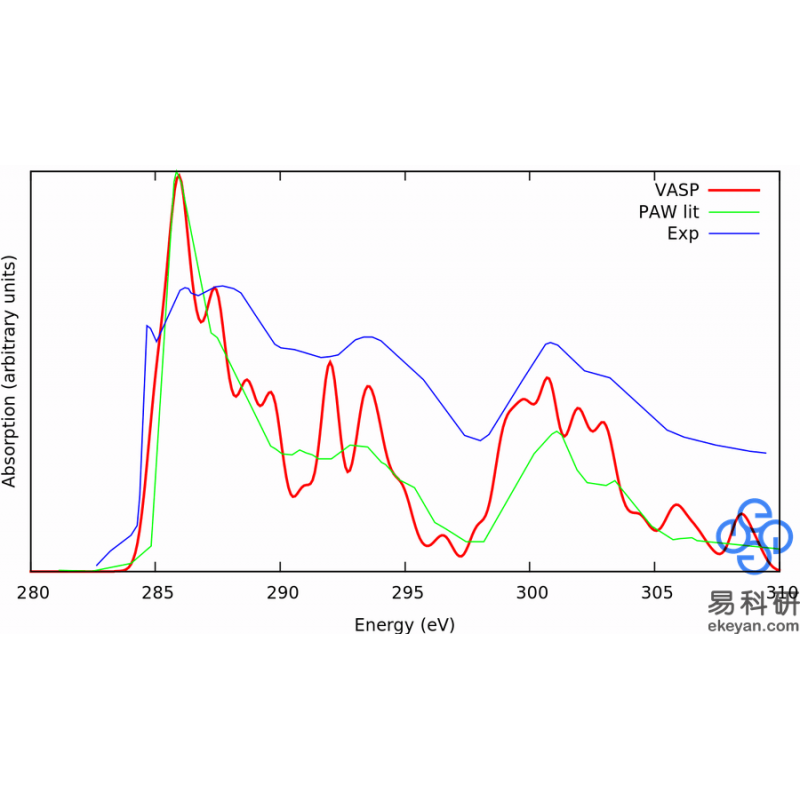
1) XANES in Diamond
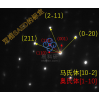
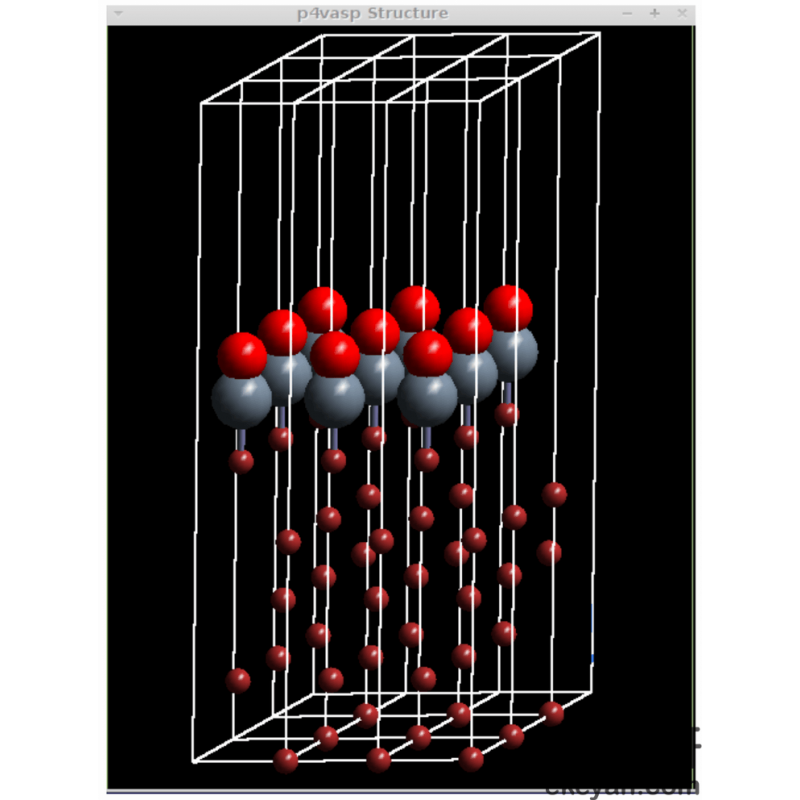
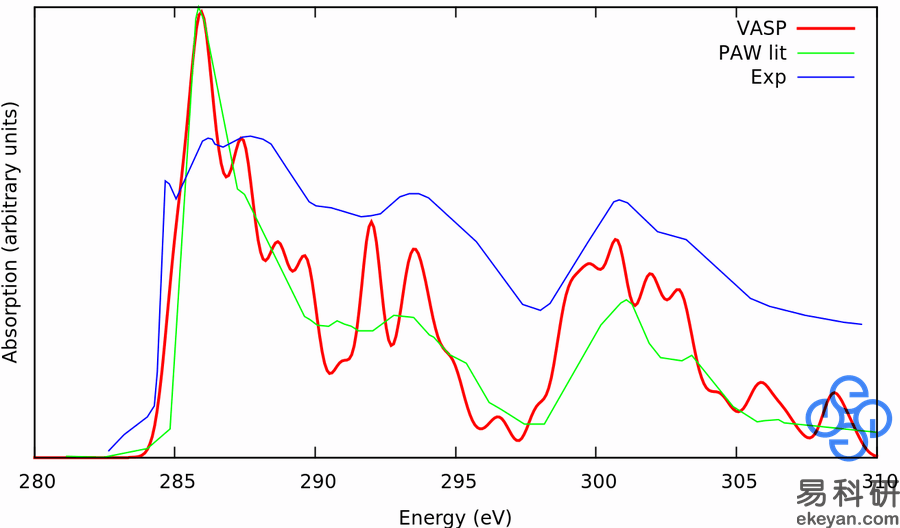 2) CO on Ni 111 surface
2) CO on Ni 111 surface
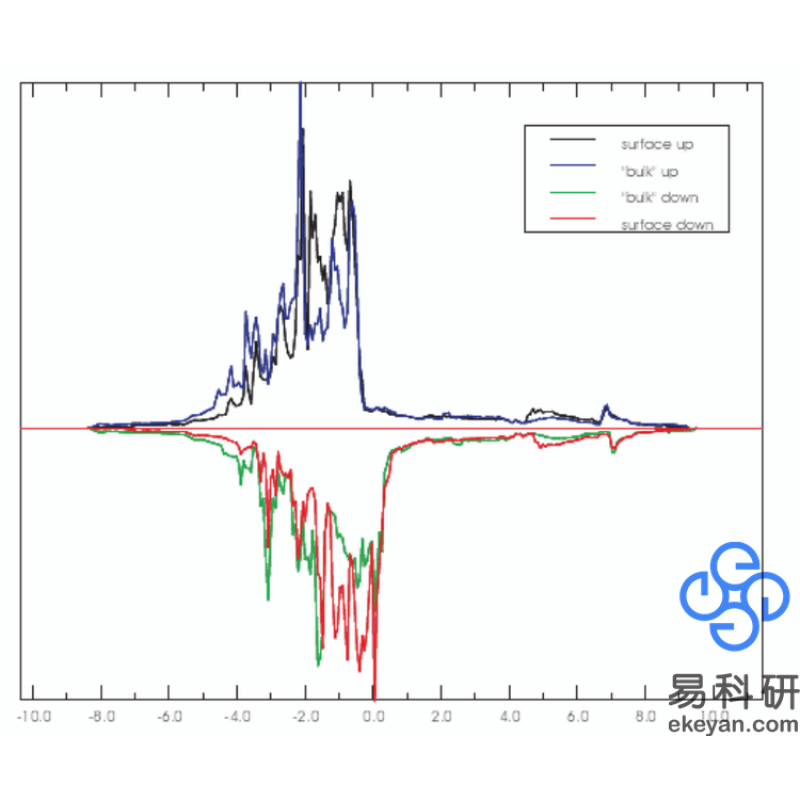
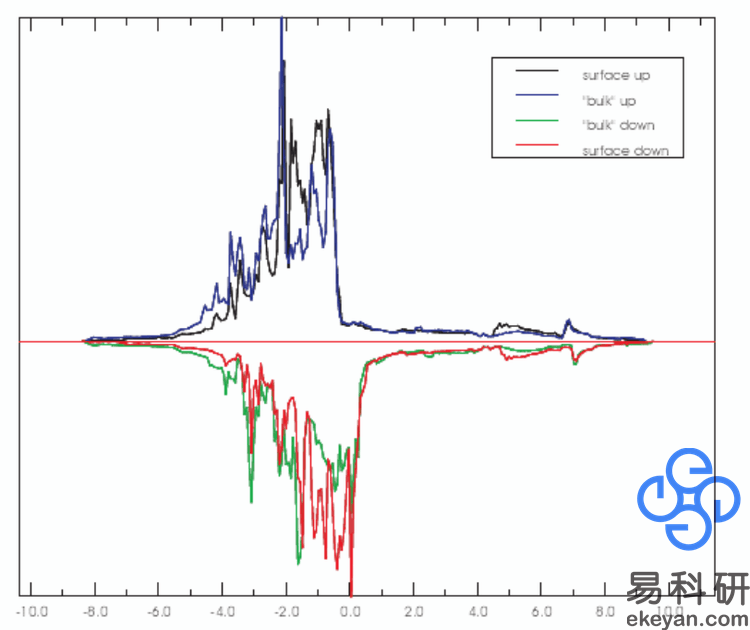
 3) Ni 100 surface DOS
3) Ni 100 surface DOS
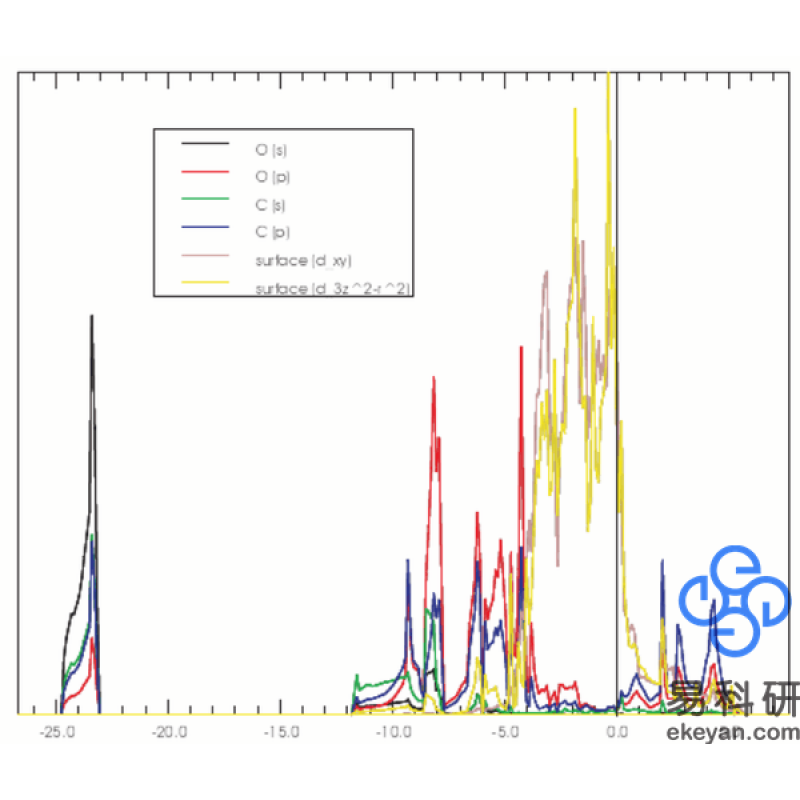
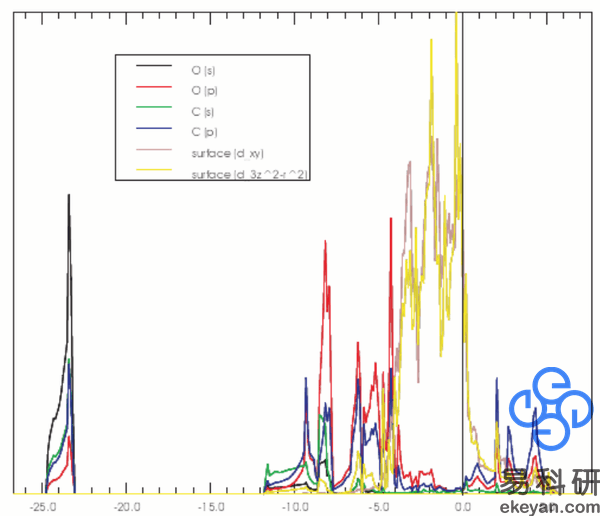
 4) Partial DOS of CO on Ni 111 surface
4) Partial DOS of CO on Ni 111 surface
主要功能
1) 采用周期性边界条件(或超原胞模型)处理原子、分子、团簇、纳米线(或管)、薄膜、晶体、准晶和无定性材料,以及表面体系和固体; 2) 计算材料的结构参数(键长,键角,晶格常数,原子位置等)和构型; 3) 计算材料的状态方程和力学性质(体弹性模量和弹性常数); 4) 计算材料的电子结构(能级、电荷密度分布、能带、电子态密度和ELF); 5) 计算材料的光学性质; 6) 计算材料的磁学性质; 7) 计算材料的晶格动力学性质(声子谱等); 8) 表面体系的模拟(重构、表面态和STM模拟); 9) 从头分子动力学模拟; 10) 计算材料的激发态(GW准粒子修正)
收费标准
根据计算具体需求评估计算工期和费用,详情请联系在线客服
Vienna Ab-initio Simulation Package(VASP)
![]() VASP是维也纳大学Hafner小组开发的进行电子结构计算和量子力学-分子动力学模拟软件包。它是目前材料模拟和计算物质科学研究中最流行的商用软件之一。VASP通过近似求解Schrödinger方程得到体系的电子态和能量,既可以在密度泛函理论(DFT)框架内求解Kohn-Sham方程(已实现了混合(hybrid)泛函计算),也可以在Hartree-Fock(HF)的近似下求解Roothaan方程。此外,VASP也支持格林函数方法(GW准粒子近似,ACFDT-RPA)和微扰理论(二阶Møller-Plesset)。VASP使用平面波基组,电子与离子间的相互作用使用模守恒赝势(NCPP)、超软赝势(USPP)或投影扩充波(PAW)方法描述
VASP是维也纳大学Hafner小组开发的进行电子结构计算和量子力学-分子动力学模拟软件包。它是目前材料模拟和计算物质科学研究中最流行的商用软件之一。VASP通过近似求解Schrödinger方程得到体系的电子态和能量,既可以在密度泛函理论(DFT)框架内求解Kohn-Sham方程(已实现了混合(hybrid)泛函计算),也可以在Hartree-Fock(HF)的近似下求解Roothaan方程。此外,VASP也支持格林函数方法(GW准粒子近似,ACFDT-RPA)和微扰理论(二阶Møller-Plesset)。VASP使用平面波基组,电子与离子间的相互作用使用模守恒赝势(NCPP)、超软赝势(USPP)或投影扩充波(PAW)方法描述
主要技术参数
1)能够对体系进行几何结构优化,获得稳定构型,包括键长、键角、晶格常数、原子位置等 2)能够计算体系的总能量及原子受力 3)能够计算体系的电子性质(波函数、能带结构、电子态密度、电荷密度等) 4)能够计算体系的状态方程、弹性常数、玻恩有效电荷、静态介电张量、压电张量、介电函数、磁矩、频率等性质 5)能够计算表面上分子或团簇的吸附能和反应的过渡态 6)能够进行DFT+U计算 7)支持非共线磁性和自旋轨道耦合计算 8)支持从头分子动力学(MD)计算,对MD的每一步都采用有效矩阵对角方案和有效Pulay混合求解瞬时电子基态,支持NVT和NVE 9)支持激发态完全含频GW计算,速度达到等离子极点模型。 10)支持LDA和GGA混合泛函包括:LDA、AM05、PBEsol、PBE、rPBE、BLYP 11)支持杂化泛函包括:HSE06、PBE0、B3LYP 12)支持metaGGA泛函包括: revTPSS、TPSS、M06-L 13)支持范德华密度泛函包括:optB86b-vdW,optB88-vdW,optPBE-vdW,rPW86-vdW2,revPBE-vdW 14)支持Screened Exchange和Hartree-Fock等非局域相互作用方法 15)采用PAW(缀加波贋势)进行量子力学计算,涵盖整个周期表内所有元素的贋势。每种元素的贋势都按照是否处理semi-core态,分为A,A_sv,A_pv和A_d,按截断能的大小,分为普通赝势A,软赝势A_s和硬赝势A_h 16)能够进行并行计算,无核数和节点限制,支持多用户同时使用