








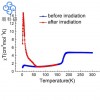































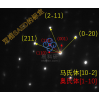








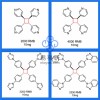
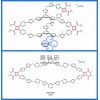

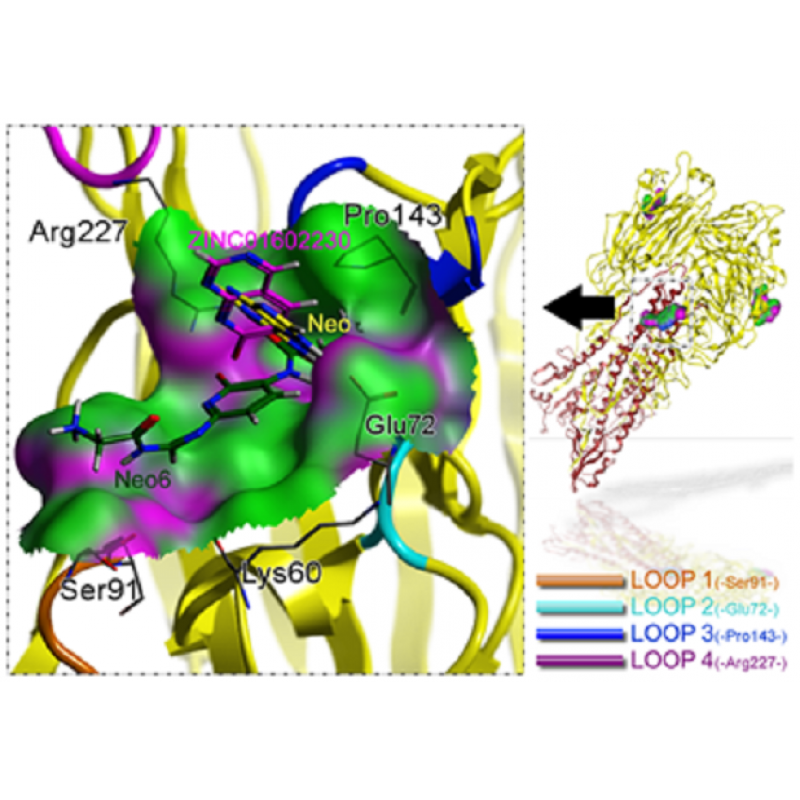
分子动力学模拟(Gromacs)
计算样例
1) 样例1
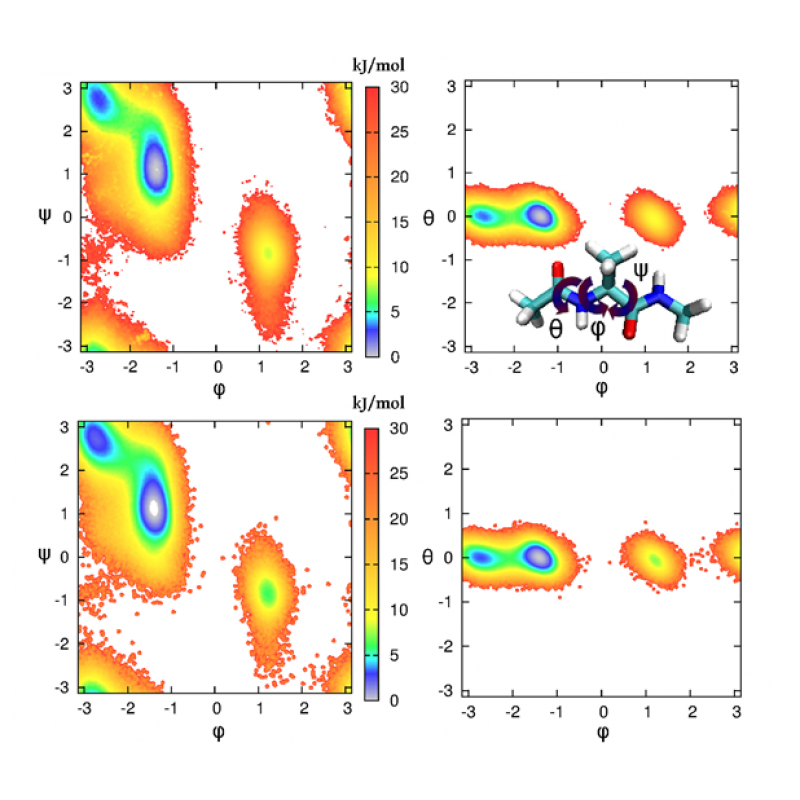
 2) ;样例2
2) ;样例2
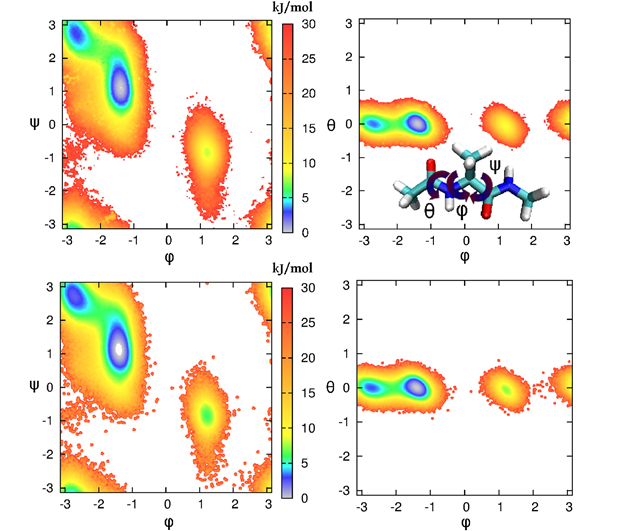
 3) ;样例3
3) ;样例3
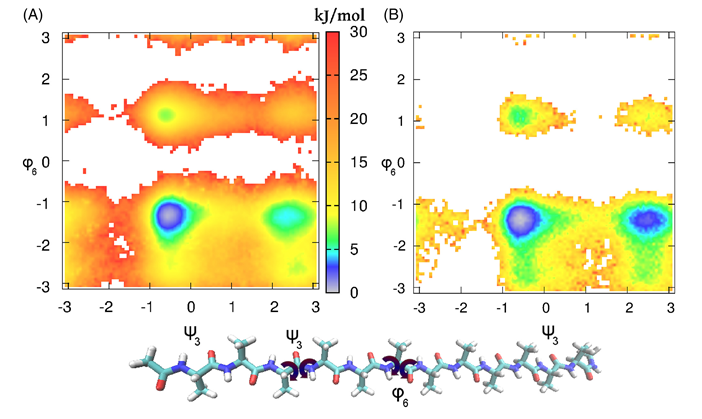
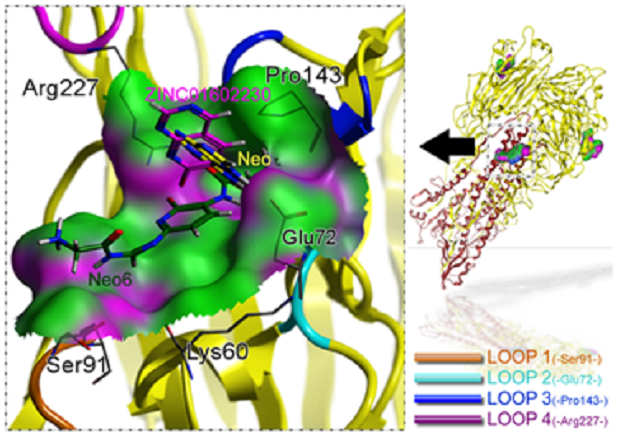 4) ;样例4
4) ;样例4
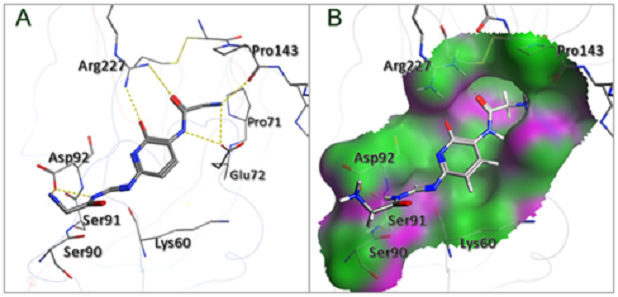
主要功能
1) 支持基本动力学相关算法,包括牛顿力学及随机动力学积分器、能量最小化、正则模式分析、模拟淬火等。支持温度及压强控制,支持基于SHAKE和P-LINCS的完全约束算法,支持多种几何约束。支持包括umbrella sampling在内的非平衡态动力学。支持FEP、essential dynamics。暂不支持const-pH 2) 内建支持AMBER、CHARMM、GROMOS及OPLS等多种常见经典力场及数种较少见力场。软件通过user tables实现对非标准函数形式的支持 3) 支持Martini粗粒化模型 4) 支持基于GBSA的隐式溶剂模型,包括三种可选的计算Bornradii的方法 5) 支持QM/MM混合动力学,对QM部分支持的计算方法包括AM1,PM3,RHF,UHF,DFT,B3LYP,MP2,CASSCF和MMVB,可对接GAMESS、Gaussian、MOPAC、Orca等量化软件
收费标准
根据计算具体需求评估计算工期和费用,详情请联系在线客服
分子动力学模拟(Gromacs)
 Gromacs是用于研究生物分子体系的分子动力学程序包。它可以用分子动力学、随机动力学或者路径积分方法模拟溶液或晶体中的任意分子,进行分子能量的最小化,分析构象等。模拟程序包包含Gromacs力场(蛋白质、核苷酸、糖等),研究的范围可以包括玻璃和液晶、到聚合物、晶体和生物分子溶液。Gromacs是一个功能强大的分子动力学的模拟软件,其在模拟大量分子系统的牛顿运动方面具有极大的优势。
Gromacs是用于研究生物分子体系的分子动力学程序包。它可以用分子动力学、随机动力学或者路径积分方法模拟溶液或晶体中的任意分子,进行分子能量的最小化,分析构象等。模拟程序包包含Gromacs力场(蛋白质、核苷酸、糖等),研究的范围可以包括玻璃和液晶、到聚合物、晶体和生物分子溶液。Gromacs是一个功能强大的分子动力学的模拟软件,其在模拟大量分子系统的牛顿运动方面具有极大的优势。
主要技术参数
1)能够对体系进行几何结构优化,获得稳定构型,包括键长、键角、晶格常数、原子位置等 2)能够计算体系的总能量及原子受力 3)能够计算体系的电子性质(波函数、能带结构、电子态密度、电荷密度等) 4)能够计算体系的状态方程、弹性常数、玻恩有效电荷、静态介电张量、压电张量、介电函数、磁矩、频率等性质 5)能够计算表面上分子或团簇的吸附能和反应的过渡态 6)能够进行DFT+U计算 7)支持非共线磁性和自旋轨道耦合计算 8)支持从头分子动力学(MD)计算,对MD的每一步都采用有效矩阵对角方案和有效Pulay混合求解瞬时电子基态,支持NVT和NVE 9)支持激发态完全含频GW计算,速度达到等离子极点模型。 10)支持LDA和GGA混合泛函包括:LDA、AM05、PBEsol、PBE、rPBE、BLYP 11)支持杂化泛函包括:HSE06、PBE0、B3LYP 12)支持metaGGA泛函包括: revTPSS、TPSS、M06-L 13)支持范德华密度泛函包括:optB86b-vdW,optB88-vdW,optPBE-vdW,rPW86-vdW2,revPBE-vdW 14)支持Screened Exchange和Hartree-Fock等非局域相互作用方法 15)采用PAW(缀加波贋势)进行量子力学计算,涵盖整个周期表内所有元素的贋势。每种元素的贋势都按照是否处理semi-core态,分为A,A_sv,A_pv和A_d,按截断能的大小,分为普通赝势A,软赝势A_s和硬赝势A_h 16)能够进行并行计算,无核数和节点限制,支持多用户同时使用